


Siguiente: Soluciones
Subir: Sesión de demostración práctica
Anterior: Soluciones
Realizamos una búsqueda BLAST(Altschul et al., 1997) con nuestra proteína problema (query
en inglés) y encontramos la proteína 1LFU,
de la cual conocemos su estructura tridimensional. A pesar de que la identidad de secuencia
entre ambas secuencias es baja, alrededor del 30%, usaremos a 1LFU como molde para hacernos
una idea de la estructura atómica de la proteína del pez, basándonos en este alineamiento:
unnamed ------KNATRESTSTLKAWLSEHRKNPYPTKGEKIMLAIITKMTLTQVSTWFANARRRLKKENKMTWTPRNRTDEEGNVYSS------
PDB1LFU MARRKRRNFNKQATEILNEYFYSHLSNPYPSEEAKEELAKKSGITVSQVSNWFGNKRIRYKKN-------IGKFQEEANIYAAKTAVTA
Puedes inspeccionar las coordenadas atómicas de 1LFU consultando el archivo
1lfu.pdb. Este archivo está en formato PDB y
contiene 3 cadenas diferentes: A y B, que forman un dúplex de ADN y P, el polipéptido.
Además necesitarás el archivo tetraodon.align
para resolver el ejercicio.
Estas son las tareas propuestas:
- completa el programa modelcomp_incompleto.pl
para implementar un algoritmo simplificado de modelado comparativo de proteínas y aplicarlo a nuestro
ejemplo. Antes de intentarlo asegúrate de que comprendes el código.
- ejecuta el programa y construye un modelo sin cadenas laterales en formato PDB.
- visualiza el modelo que hayas construído con algún programa gráfico como rasmol.
- calcula la estructura secundaria del modelo obtenido usando la interfaz web del programa
STRIDE. Deberás
guardar el archivo stride.out.
- a partir de la estructura secundaria obtenida (repasa la sección 2.5), haz una
estima de la velocidad de plegamiento de esta proteína, usando la expresión (medida en
 ):
):
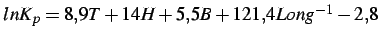
- estima el número de conformaciones diferentes que podría tener esta secuencia y qué
tiempo máximo tomaría explorarlas todas, de acuerdo con el razonamiento de Levinthal.
Siguiente: Soluciones
Subir: Sesión de demostración práctica
Anterior: Soluciones
Bruno Contreras Moreira
2006-11-08