![\begin{figure}\begin{center}
\includegraphics[width=0.8\textwidth]{cluster}\end{center}\end{figure}](img1.png) |
cursos 2005/2007
Licenciatura en Ciencias Genómicas, UNAM
©Bruno Contreras-Moreira and Heladia Salgado
Con aportaciones de Cei Abreu-Goodger
Programa de Genómica Computacional
Centro de Ciencias Genómicas, UNAM
62210 Cuernavaca, Morelos
México
Este curso es parte de la licenciatura de Ciencias Genómicas de la Universidad Nacional Autónoma de México y está pensado para alumnos y alumnas que ya tengan cierta experiencia en bioinformática.
Podéis mandarme comentarios o sugerencias através de la
dirección de correo contrera_arroba_ccg.unam.mx.
A pesar de que los sistemas operativos Windows dominan el mundo, puede decirse que en el ámbito de la bioinformática el rey es UNIX, normalmente en forma de linux. En esta parte del curso queremos mostrar algunas utilidades muy poderosas del mundo UNIX aplicables a la bioinformática.
Este tutorial tiene como objetivo capacitar al estudiante en el uso de comandos avanzados de UNIX en tareas bioinformáticas, viendo temas como scripts Perl/shell, combinaciones de comandos.
Archivos de datos usados en los ejemplos:
ECK12.gene , lista de genes de Escherichia coli K-12
genes_db.GeneSeq.txt , secuencias de los genes de Escherichia coli K-12
NetWorkSet.txt , Red de regulación de Escherichia coli K-12
man
ls lista el contenido de un directorio
cat/more/less despliega el contenido de un archivo
mkdir crea un directorio
rmdir borrar un directorio
rm borrar un archivo
cp copiar un archivo
mv mover o renombrar un archivo
wc contar caracteres, palabras y l{ineas de un archivo
pwd desplegar el path del directorio actual
| Conecta procesos que se ejecutan consecutivamente >> Redirecciona la salida estándar < Redirecciona la entrada estándar
sort Ordena las líneas de un archivo.
Algunas opciones:
Ejemplos:
$ sort -k3 ECK12.gene > l
$ sort -k4 -n l | more
$ cut -f1 NetWorkSet.txt | sort -u | more
cut Extrae campos seleccionados de cada línea de un archivo.
Algunas opciones:
Ejemplos:
$ cut -f2,3 ECK12.gene
$ cut -f3,8 ECK12.gene
$ cut -f3 genes_db.GeneSeq.txt | cut -c1-3 | more
$ cut -f3 genes_db.GeneSeq.txt | perl -ne 'if ($_ =~ /(...)$/) { print "$1\n"; }' - | more
uniq Reporta o filtra líneas consecutivas repetidas.
Algunas opciones:
Ejemplos:
$ cut -f1 NetWorkSet.txt | sort | uniq | more
$cut -f6 ECK12.gene | sort | uniq -c
grep Busca un patrón en un archivo e imprime las lineas donde el patrón fue encontrado.
Algunas opciones:
Ejemplos:
$ grep -w LexA NetWorkSet.txt | cut -f4 | sort -u
$ grep -w sodA NetWorkSet.txt | cut -f1 | sort -u
sed
Es un editor de textos, Veamos como ejemplo como substituir los simbolos + - +- por activador, represor y dual:
$ sed 's/+-/dual/; s/+/activator/; s/\-/repressor/;' NetWorkSet.txt | more
perl Practical Extraction and Report Language
Algunas opciones en linea de comando:
$_ es almacenado en @F
Veamos algunos ejemplos que manipulan Feature maps en este formato:
$ perl -a -ne 's/reverse/R/; s/forward/F/; print "$F[2]\tORF\t$F[1]\t$F[5]\t$F[3]\t$F[4]\n";' ECK12.gene >Escherichia_coli_K12.genes
$ perl -a -ne ' if(($F[3]>=2000000) && ($F[3]>=3000000)) { print; } ' ECK12.gene | wc
$ grep -w LexA NetWorkSet.txt | cut -f4 | sort -u | infer-operon -org Escherichia_coli_K12 -return operon
La utileria make determina que partes de un programa extenso necesitan ser recompilados. Tambien ayuda a organizar y ejecutar un conjunto de tareas.
Ayuda en la Web:
Make necesita de un archivo llamado makefile que describe cómo y qué compilar o ejecutar. Por defecto make busca archivos que se llaman Makefile, pero se le pueden pasar archivos con otros nombres con la opción -f.
Un makefile consiste de reglas de la siguiente forma:
target ... : dependencies ...
command
...
Como ejemplos veamos los makefiles contenidos en MakeF/ejemplo_C (compilación) y en MakeF/makefile
(ejecución de tareas).
Resultados esperados:
987 + 157 +- 789 - 3 ?
AcrR - 3 Ada + 2 Ada +- 2 AdiY + 1 AgaR - 11 AllR - 10
żQué es un cluster o granja? Es un grupo de computadoras, normalmente PCs por su relativamente reducido precio, que se conectan en forma de red a un nodo maestro que reparte las tareas que hay que realizar entre ellas. Cada computadora en esta red es un nodo, destacando el nodo maestro, que realiza la función de interfaz entre los usuarios y los nodos de cómputo. La arquitectura de los clusters permite normalmente que todo siga funcionando bien si se rompe cualquier nodo, excepto el maestro.
El nodo maestro de un cluster ejecuta un programa servidor que gestiona los recursos del cluster, normalmente por medio de colas. Dos ejemplos muy populares de este software son torque (basado en PBS) y Sun Grid Engine. Una de las tareas de este tipo de aplicaciones es conocer en cada instante qué recursos están disponibles y cuáles están ocupados, como podemos ver en esta liga para el cluster kayab del Centro de Ciencias Genómicas.
żPara qué sirve un cluster? Para realizar tareas computacionalmente costosas, que tardarían días en tu máquina personal, en tiempos reducidos.
Normalmente, un usuario bioinformático desarrolla sus programas en su máquina personal y los prueba con ejemplos sencillos para ir depurando el código. Sólo una vez que el programa hace lo que se desea, normalmente, se ejecuta sobre ejemplos complejos, por ejemplo un genoma completo.
Supongamos que tenemos que anotar un genoma de 10000 genes. Eso implicaría ejecutar al menos 10000 veces BLAST, que en una máquina personal costaría un total de 100 horas, por poner un ejemplo (serían 36s por cada búsqueda BLAST). Si tenemos a nuestra disposición una cluster de 100 procesadores, podríamos hacer el mismo trabajo en aproximadamente una hora. Para ello necesitaríamos escribir un programa similar a éste y ejecutarlo en el nodo maestro:
#!/usr/bin/perl -w
# programa que lanza muchos BLASTs
use strict;
my $qsubEXE = "/opt/torque/bin/qsub";
my $BLASTEXE = "/usr/bin/local/blast/bin/blastpgp";
my $baseDIR = "/home/contrera/kayab/mydir/";
#...
foreach $seq (@genome)
{
# crea archivo FASTA temporal con secuencia para BLAST
my $infile = $baseDIR . $seq . ".inblast";
open(INTMP,">$infile");
print INTMP ">$seq\n$seq\n";
close(INTMP);
# envía trabajo BLAST con esta secuencia
my $outfile = $baseDIR . $seq . ".outblast";
my $PBSoutfile = $baseDIR . $seq . ".pbsout";
my $qsubcommand = " -l nodes=1 -N n$seq -q default -j oe -o $PBSoutfile -S /bin/csh";
$qresult = `$qsubEXE $qsubcommand <<TRABAJO
cd $baseDIR
$BLASTEXE -i $infile -e 0.00005 -o $outfile
TRABAJO`;
# borra lo que no necesitemos...
}
Entonces, en definitiva, necesitaremos:
$ ssh usuario@kayab
żCómo sabemos si nuestro script ha funcionado? Para eso usamos el comando qstat desde el nodo
maestro. Si queremos saber en qué estado están nuestros trabajos, con más detalle, podemos invocar: qstat -n -u usuario.
Si queremos eliminar trabajos usaremos el comando qdel proceso1 proceso2 ... procesoN.
Como siempre en UNIX, podemos conocer otras maneras de ejecutar estos comandos por medio de:
man qstat man qsub man qdel
LaTeX es un lenguaje de descripción de documentos que permite la edición profesional de textos. Un documento escrito en LATEX es pasado a un intérprete que a su vez genera la versión final. żQué interés tiene LATEX cuando se pueden usar procesadores de texto más sencillos como OpenOffice o Word?
En esta sección veremos como podemos usar el mismo texto formateado con LATEX para generar
un documento web navegable con ligas internas (usando
latex2html) y un documento
imprimible de alta calidad en formato PDF.
Para ello trabajaremos con el ejemplo contenido en /home/bioinfo/latex, obtenido de
acá. La idea es inspeccionar
el código, escribir código propio y aprender a compilarlo. Fíjate en el contenido de los diferentes
Makefile y a ver si averiguas cómo:
Nedit o vim
dvipdf
.tex, modificando algún Makefile
Con el fin de mantener una colección de artículos en formato Bibtex podríamos usar algunas herramientas web como éstas:
No lo veremos aquí, pero es posible también generar presentaciones de alta calidad basadas en documentos LATEX , usando la clase beamer.
El analisis de datos de manera estadística y gráfica llega a ser una necesidad para la interpretación biológica. R proporciona un ambiente sumamente poderoso y fácil para estas tareas, y que incluso existen paquetes de analisis completos en diversas areas de la genómica y que pueden ser importados a R.
Bioconductor es un software "open source" para análisis de datos genómicos.
Bajar e instalar desde http://www.r-project.org
Manuales: http://cran.r-project.org/manuals.html
El simbolo % indica el prompt de Unix y > el prompt de R.
Desde Unix:
% R # iniciar R > q(); # salir del programa.
Windows: Busca el "shortcut" de R para iniciar.
> help(mean) ## get help for the function > ?mean > # : everything after # in the line is remark. i.e. R will ignore it.
> ## Note: R is case-sensitive.
+ ## este prompt en R indica que aun no esta complete el commando, y dam as lineas
para terminarlo.
> source('command.R') # comandos almacenados en un archivo
> x
> y
> sink('result.list') # todas las salidas de comandos las manda a un archivo
> sink() # restablece las salidas a la pantalla.
> objects() # display the names of the objects which are currently stored within R
> rm(x,y,z,temp) # remove objects
> (2.3 + 4.3 * 3) ^ 0.7 / 2 # orden: ^, * / , + -
Operador de asignación <-
> x <- 6 > 8 -> x
Nombrando objetos: Letras y números son permitidos ( pero el primer caracter tiene que ser una letra), el "." También es permitido.
Objetos, sus modos y atributos. Las variables o entidades usadas en R son conocidas como objetos, y cada uno de los valores son sus componentes. Cada objeto tiene un tipo de dato o modo como son numeric, complex, logical, character, list, function. Cada objeto tiene ciertas propiedades o atributos como son el tamano, el modo, etc..
function mode Get or set the type or storage mode of an object.
> x<-1:5 ## mode: numeric, complex, logical, character, list, function > x > mode(x) > x<-as.character(x) > x > mode(x)
function Length:
Length of an Object.
> x<-numeric() ## make it an empty vector with length indefinite > x[3]<-7 ## now x has length 3 with the third element equals 7.
function c Combina valores en un vector o lista.
> x <- c(10.4, 5.6, 3.1, 6.4, 21.7) ## assign a vector of numbers to object x.
> z <- c(1,7:9) ## rango de valores
> a <- c("Hello! World!") ## assign a character string to object a
Vectores Aritméticos
Operadores aritmeticos: +, -, *, /, ^, log, exp, sin, cos, tan, sqrt, min, max, length, sum, mean, var, sd, order, sort, rank > x <- c(1,2,3) > y <- c(5, 6, 7) > x + y > y<- 5 > x + y
ejemplo de manejo de vectores numericos
> x<- c(1, 2, NA, 3, 4) ## NA is missing value > is.na(x) > length(x) > mean(x) ## the command won't run. The default parameter for mean doesn't allow missing value. > help(mean) > mean(x, na.rm=T) ## set the parameter so the mean function allows missing value > x<- c(10.4, 5.6, 3.1, 6.4, 21.7) > var(x) # varianza > sum( (x-mean(x))^2 ) / (length(x)-1)
Generando secuencias:
> x<-1:10 > x<-seq(-5, 5, by=.2) > x<-rep(1:3, times=5) > x<-rep(1:3, each=5)
Vectores de caracteres
Funcion paste. Concatena vectores despues de convertirlos a caracter.
> paste(1:12) # same as as.character(1:12)
> paste("Today is", date())
> x<-paste("Affy", 1:5, sep="")
> x<-paste(c("X", "Y"), 1:5, sep="_")
Vectores Logicos:
> x<- (-5)+(0:15) > x>0 > x>0 & x<=5
Seleccion de valores de un vector:
> x<- (-5)+(0:15) > x > x[3:6] > x[x>0 & x<=5] > x[-(3:6)] ## - eliminar elementos del rango indicado > x[-(x>0 & x<=5)] > x[1:3]<-100:102 ## modify the first three number > x[(x>0 & x<=5)]<-999 ## modify the data satisfying the selection
> x <- 1:20 > dim(x) <- c(4,5) ## convert a vector to a 4X5 array > x[c(2,4),] ## print the second and the fourth row´ > x[,c(2,4)] ## print the 2,4 columns > x<-array(1:20, dim=c(4,5)) > x > i<-array(c(1:3, 3:1), dim=c(3,2)) > i > x[i]<-0 ## asigna 0 a las posiciones 1,3; 2,2; y 3,1 de x > x > y<-cbind(x, nrow(x):1) ## pega x y un vector nrow para generar y -:1 toma etiquetas del renglon > y<-rbind(x, ncol(x):1) ## row-wise bind x and vector ncol(x):1
funcion matrix
Crea una matrix desde un conjunto de valores dados.
matrix(data = NA, nrow = 1, ncol = 1, byrow = FALSE, dimnames = NULL)
> ## assign a matrix to object ma
> ma <- matrix(c(1,2,3, 11,12,13), nrow = 2, ncol=3, byrow=TRUE,
dimnames = list(c("row1", "row2"), c("C.1", "C.2", "C.3"))) ##default
byrow=FALSE
Es un objeto que consiste de una coleccion de objetos conocidos como componentes de la lista.
> Lst <- list(name="Fred", wife="Mary", no.children=3, child.ages=c(4,7,9)) > Lst[[1]] > Lst$name > Lst[[4]][2]
> accounts<-data.frame(state=c("PA", "CA", "MA"), income=c(1000, 1100, 900),
married=c(T, F, F))
> attributes(accounts)
Random number generator:
Function rnorm, dnorm, pnorm, qnorm. Dnorm gives the density, 'pnorm' gives the distribution function, 'qnorm' gives the quantile function, and 'rnorm' generates random deviates.
> a<-rnorm(1000, 5, 1) ## get 1000 samples from normal distribution, means 5 and stan deviation 1. > hist(a, nclass=30) > pnorm(1.96) > qnorm(0.975)
other functions:
Rnorm, runif, rt, rchisq, rexp, rweibull, rbeta, rgamma
Function scan
> list_data <- scan('test_list.dat')
> list_data[8]
Funcion read.table Reads a file in table format and creates a data frame from it
> HousePrice <-read.table(´/home/bioinfo/cursoFeb_Jun_2007/R_sesison/data/houses.data´)
> HousePrice
> HousePrice <-cbind(HousePrice, name=c('c1','c2','c3','c4','c5'))
funcion write.table
Escribe el contenido de una variable a un archivo en disco.
> write.table(data,'houses_v2.data') ## write out data to the file > write.table(data, 'houses_v2.data', sep="\t", quote=F) ## write out data without quotation mark and with tab delimited
> HousePrice <-read.table(´/home/bioinfo/cursoFeb_Jun_2007/R_sesison/data/houses.data´) > plot(data$area, data$price) ## plot area versus price
Prepare a graphic device for drawing
> jpeg( 'houseInfo.jpg', width = 600, height = 480)
> plot(data$area, data$price, xlab="area", ylab="price") ## draw on the graphic device
> title("house information") ## add title to the plot
> dev.off() ## close the graphic device and complete drawing
> png('houseInfo.png', width = 480, height = 600)
> plot(data$area, data$price, xlab="area", ylab="price")
> title("house information")
> dev.off()
## add from 1 to 100
a<-0 ## initialize a
for(i in 1:100) ## run the loop from i=1 to i=100
{
a<-a+i ## each time add i to a
}
a ## the output of a
## add all positive numbers in the vector
x<-c(2.1, 9.3, -1.2, 2.43, -4.32, -7.3, 5.29)
a<-0
for(i in 1:length(x))
{
if(x[i]>0) a<-a+x[i] ## add to a if x[i]>0
}
## note: a simpler alternative, a<-sum(x[x>0])
absolute.sum<-function(n) ## a function that adds from -n to n
{
a<-0
for(i in -n:n)
{
if(i>=0) a<-a+i ## if i is positive, add i to a
else a<-a+(-i) ## if i negative, add the absolute value -i to a
}
return(a)
}
> absolute.sum(100)
Tip: looping is very expensive in R. Always avoid looping if possible
absolute.sum1<-function(n) ## this is another version of faster implementation
{ return(sum(abs(-n:n))) }
absolute.sum(1000000) ## this will take a while
absolute.sum1(1000000) ## this will take less than a second
Install Bioconductor:
> source("http://www.bioconductor.org/getBioC.R")
> ## this will download the getBioC functionality into your R session.
> getBioC("affy") ## this will install "affy" and "exprs" packages
> ### Load a package:
> library("affy") ## load the "affy" library
Web package page:
http://www.bioconductor.org/repository/release1.5/package/html/
Biobase Object-oriented representation and manipulation of genomic data (S4 class structure).
Biostrings Class definitions and generics for biological sequences along with pattern matching algorithms
convert Define coerce methods for microarray data objects
ctc Tools for export and import of Tree and Cluster to other programs
DynDoc Functionality to create and interact with dynamic documents, vignettes, and other navigable documents.
exprExternal Provides a subClass of exprSet from the Biobase package which implements the same features but uses an external matrix object with some arbitrary external storage.
externalVector Basic class definitions and generics for external pointer based vector objects for R
Icense Many functions for computing the NPMLE for censored and truncated data.
reposTools Tools for dealing with file repositories and allow users to easily install, update, and distribute packages, vignettes, and other files.
Ruuid Creates Universally Unique ID values (UUIDs) in R
annaffy Functions for handling data from Bioconductor Affymetrix annotation data packages. Produces compact HTML and text reports including experimental data and URL links to many online databases.
marray Diagnostic plots and normalization for cDNA microarray data.
matchprobes Tools for sequence matching of probes on arrays
vsn Calibration and variance stabilizing transformations for both Affymetrix and cDNA array data.
edd Expression density diagnostics: graphical methods and pattern recognition algorithms for distribution shape classification.
factDesign Provides a set of tools for analyzing data from factorial designed microarray experiments. The functions can be used to evaluate appropriate tests of contrast and perform single outlier detection
genefilter Tools for sequentially filtering genes using a wide variety of filtering functions. Example of filters include: number of missing value, coefficient of variation of expression measures, ANOVA p-value, Cox model p-values. Sequential application of filtering functions to genes
globaltest Testing globally whether a group of genes is significantly relatedto some clinical variable of interest.
gpls Classification using generalized partial least squares for two-group and multi-group
limma Linear models for microarray data
RMAGEML Used to handle MAGE-ML documents in Bioconductor
MeasurementError.cor Two-stage measurement error model for correlation estimation with smaller bias then the usual sample correlation
multtest Multiple testing procedures for controlling the family-wise error rate (FWER) and the false discovery rate (FDR). Tests can be based on t- or F-statistics for one- and two-factor designs, and permutation procedures are available to estimate adjusted p-values.
pamr Some functions for sample classification in microarrays
ROC Receiver Operating Characteristic (ROC) approach for identifying genes that are differentially expressed int two types of samples.
siggenes Identifying differentially expressed genes and estimating the False Discovery Rate with both the Significance Analysis of Microarrays and the Empirical Bayes Analyses of Microarrays
splicegear A set of tools to work with alternative splicing
annotate Associate experimental data in real time to biological metadata from web databases such as GenBank, LocusLink and PubMed. Process and store query results. Generate HTML reports of analyses.
AnnBuilder Assemble and process genomic annotation data, from databases such as GenBank, the Gene Ontology Consortium, LocusLink, UniGene, the UCSC Human Genome Project.
Data packages XML and R annotation data packages, providing mappings between different probe identifiers (e.g. Affy IDs, LocusLink, PubMed)
Resourcer This package allows user either to read an annotation data file from TIGR Resourcerer as a matrix or convert the file into a Bioconductor annotation data package using the AnnBuilder package.
SNPtools Provides bindings to the XML RPC services of chip.org::SNPper
affylmGUI A Graphical User Interface for affy analysis using the limma Microarray package by Gordon Smyth.
geneplotter Graphical tools for genomic data, for example for plotting expression data along a chromosome or producing color images of expression data matrices.
hexbin Binning functions, in particular hexagonal bins for graphing.
limmaGUI A Graphical User Interface for the limma Microarray package
tkWidgets Widgets in Tcl/Tk that provide functionality for Bioconductor packages.
webbioc An integrated web interface for doing microarray analysis using several of the Bioconductor packages. It is intended to be deployed as a centralized bioinformatics resource for use by many users. (Currently only Affymetrix oligonucleotide analysis is supported.)
widgetTools Tools for creating Tcl/Tk widgets, i.e., small-scale graphical user interfaces.
RBGL A package that creates an interface between the graph package and the Boost graph libraries, allowing for fast manipulation of graph objects in R.
Rgraphviz Provides an interface with Graphviz for plotting graph objects in R.
SNAData Data from Wasserman and Faust (1999) "Social Network Analysis"
#########################################################################
#### Practica de LCG para la materia de BioInformatica aplicada ####
#### Contiene: instrucciones para la instalacion completa de ####
#### Bioconductor y un ejemplo de analisis de un data set de 8 ####
#### Affymetrixarrays. Seimplementan y comparan MAS 5.0, ####
#### GC-RMA (PM-MM model) and GC-RMA (PM only model) ####
#### Rosa Maria Gutierrez 15/02/07 ####
#########################################################################
## Datos bajados de http://www.affymetrix.com/analysis/download_center2.affx
## HUMAN GENOME U95 DATA SET
## instalar Bioconductor
source("http://www.bioconductor.org/biocLite.R")
biocLite()
getBioC("pkg1") ## Para instalar paquetes sueltos
biocLite(c("pkg1", "pkg2"))## Para instalar paquetes sueltos
## Cargar librerias
library("affy") ## cargar libreria affy package
library("matchprobes") ## la libreria gcrma necesita la libreria matchprobes
library("gcrma") ## cargar libreria gcrma package
dataDir<-"Affymetrixarrays/data"
workingDir<-"Affymetrixarrays"
fileNames<-dir(dataDir)
fileNames<-paste(dataDir, "/", fileNames, sep="")
LatinData<-ReadAffy(filenames=fileNames)
## 8 arreglos para el analisis: "1521m99hpp_av06.CEL" "1521n99hpp_av06.CEL"
"1521o99hpp_av06.CEL" "1521p99hpp_av06.CEL"
## "1521q99hpp_av06.CEL" "1521r99hpp_av06.CEL" "1521s99hpp_av06.CEL"
"1521t99hpp_av06.CEL". Los 4 primeros son un juego de replicas
# y los otros cuatro son otro juego de replicas
## Calcular medidas de expresion por MAS 5.0
LatinData.mas5<-expresso(LatinData, pmcorrect.method="mas", bgcorrect.method="mas",
summary.method="mas")
LatinData.mas5.norm<-affy.scalevalue.exprSet(LatinData.mas5)
write.table(exprs(LatinData.mas5.norm), paste(workingDir,
"\\LatinData_mas5_norm.txt", sep=""), sep="\t")
## Calcular medidas de expresion por gcRMA (PM-only)
LatinData.gcrma<-gcrma(LatinData, type="affinities")
write.table(exprs(LatinData.gcrma), paste(workingDir, "\\LatinData_gcrma_noMM.txt",
sep=""), sep="\t")
## Calcular medidas de expresion por gcRMA (contain MM)
LatinData.gcrma.MM<-gcrma(LatinData, type="fullmodel")
write.table(exprs(LatinData.gcrma.MM), paste(workingDir, "\\LatinData_gcrma_MM.txt",
sep=""), sep="\t")
#################
## Definicion de funciones en R
evaluate<-function(exprs.all, names)
{
par(mfrow=c(3,2))
for(i in 1:length(exprs.all))
{
a<-matrix(NA, nrow(exprs.all[[i]]), 2)
a[,1]<-apply(exprs.all[[i]], 1, mean, na.rm=TRUE)
a[,2]<-apply(exprs.all[[i]], 1, sd, na.rm=TRUE)
plot(c(0, 12), c(0, 3), type="n", xlab="mean log intensities", ylab="sd log
intensities")
points(a[,1], a[,2], pch=".")
title(names[i])
}
}
evaluate1<-function(exprs.all, names)
{
for(i in 1:length(exprs.all))
{
a<-cor(exprs.all[[i]], use="pairwise.complete.obs")
print(paste(names[i], ":", mean(a[a<1])))
}
}
## Obtener los valores de expresion de las primeras 4 replicas
LatinData.mas5.norm.matrix<-log2(exprs(LatinData.mas5.norm)[,1:4])
LatinData.gcrma.matrix<-exprs(LatinData.gcrma)[,1:4]
LatinData.gcrma.MM.matrix<-exprs(LatinData.gcrma.MM)[,1:4]
## Graficar evaluaciones.
jpeg(filename=paste(workingDir, "\\eval_replicate_1_new.jpg", sep=""), width=1200,
height=1024)
exprs.all<-list(LatinData.mas5.norm.matrix, LatinData.gcrma.matrix,
LatinData.gcrma.MM.matrix)
evaluate(exprs.all, c("MAS5","GCRMA (no MM)", "GCRMA (MM)"))
dev.off()
evaluate1(exprs.all, c("MAS5", "GCRMA(no MM)", "GCRMA(MM)"))
## print out of evaluate1
##[1] "MAS5 : 0.893039182394166"
##[1] "GCRMA(no MM) : 0.9992816227403"
##[1] "GCRMA(MM) : 0.998767869268427"
## Obtener los valores de expresion de las primeras siguentes 4 replicas
LatinData.mas5.norm.matrix<-log2(exprs(LatinData.mas5.norm)[,5:8])
LatinData.gcrma.matrix<-exprs(LatinData.gcrma)[,5:8]
LatinData.gcrma.MM.matrix<-exprs(LatinData.gcrma.MM)[,5:8]
## Plot evaluation figure again.
jpeg(filename=paste(workingDir, "\\eval_replicate_2_new.jpg", sep=""), width=1200,
height=1024)
exprs.all<-list(LatinData.mas5.norm.matrix, LatinData.gcrma.matrix,
LatinData.gcrma.MM.matrix)
evaluate(exprs.all, c("MAS5","GCRMA (no MM)", "GCRMA (MM)"))
dev.off()
evaluate1(exprs.all, c("MAS5","GCRMA(no MM)", "GCRMA(MM)"))
## print out of evaluate1
##[1] "MAS5 : 0.900234102618892"
##[1] "GCRMA(no MM) : 0.999446021907957"
##[1] "GCRMA(MM) : 0.998953186433171"
#########################################################################
#### Practica de LCG para la materia de BioInformatica aplicada ####
#### Contiene: intrucciones para la ejecucion de ejemplos ####
#### includios en bioconductor utiles para visualizar problemas ####
#### en microarreglos ####
#### Rosa Maria Gutierrez 15/02/07 ####
#########################################################################
## cargar libreria de Affymetrix
library ("affy")
## Instalar librerias de ejemplos
#source("http://www.bioconductor.org/getBioC.R")
#getBioC("SpikeInSubset")
#getBioC("affydata")
#getBioC("affycomp")
## instruccion util: Leer archivos CEL del directorio actual
# Data<- ReadAffy()
## Cargar libreria del ejemplo de Bioconductor, para visualizar datos
library("SpikeInSubset")
data("spikein95") ## llamar los datos contenidos en la libreria
## Graficas para visualizar la distribucion de los datos en los microarreglos
hist(spikein95) ## hacer un historgrama con los datos
boxplot(spikein95) ## hace una grafica de caja y bigotes
## Cargar libreria del ejemplo de Bioconductor, diluciones
library ("affydata")
data(Dilution)
pm(Dilution, "1001_at")[1:3, ] ## ver un pedazo de vector de pm
mm(Dilution, "1001_at")[1:3, ] ## ver un pedazo de vector de mm
## Graficar intensidades entre probes
matplot(pm(Dilution, "1001_at"), type ="l", xlab ="Prob No." , ylab="PM Probe
intensity")
## Graficar intensidades entre arrays
matplot(t(pm(Dilution, "1001_at")), type ="l", xlab ="Array No.", ylab="PM Probe
intensity")
## accesar informacion fenotipica
pData(Dilution)
data(rma.assessment)
data(mas5.assessment)
affycompPlot(rma.assessment$MA)
affycompPlot(rma.assessment$Signal,mas5.assessment$Signal )
En el ámbito de la biología conocemos diferentes tipos de polímeros que se pueden representar en forma de secuencias o cadenas de caracteres, como las proteínas y los ácidos nucléicos. Esta forma de representación consiste en identificar cada monómero del polímero por medio de una letra, formando así una cadena de letras o caracteres que muestran la secuencia propia del polímero.
El uso de secuencias tiene ventajas e inconvenientes. Entre las ventajas destacamos la simplificación y la facilidad de manipulación por medios computacionales. Sin embargo, perdemos información que puede ser importante. Por ejemplo, al escribir la secuencia de aminoácidos de una proteína no estamos diciendo nada acerca de su estructura tridimensional.
Un ejemplo de secuencia biológica sería el principio del gen de la insulina de la mosca
Drosophila melanogaster:
5' atgtttagcc agcacaacgg tgcagcagta 3'
donde ![]() es adenina,
es adenina, ![]() timina,
timina, ![]() citosina y
citosina y ![]() guanina.
guanina.
żQué es un alineamiento?
Un alineamiento es una comparación que hacemos entre 2 ó más secuencias con las siguientes condiciones:
żQué significado tiene un alineamiento?
La respuesta depende del contexto. Dentro del análisis de secuencias
biológicas, los alineamientos se utilizan como predictores de función, estructura o evolución. Si una secuencia A de función
desconocida se parece a otra B que sabemos es una enzima de membrana, podemos predecir con cierto margen de
error que A será también una enzima de membrana. El alineamiento de A con B será entonces esencial para
decidir cuánto se parecen A y B. Estas predicciones son posibles porque sabemos que los seres vivos han reutilizado
masivamente sus genes y proteínas y que genes similares, en organismos tan distantes como las levaduras y los seres
humanos, tienen funciones similares.
Ejemplo de alineamiento de dos secuencias de ADN con 2 gaps, observa que no todas las letras alineadas son idénticas:
aagtaaa-agcaggacggggcagcc-
atgtttacagcacaacggtgcagcag
Los algoritmos de alineamiento podrían ubicarse dentro de la familia de algoritmos útiles para la manipulación de cadenas de caracteres (Gusfield, 1997), pero aquí vamos a verlos siempre dentro del contexto biológico.
Los algoritmos de alineamiento en nuestro contexto normalmente usan matrices de pesos o sustitución para evaluar el emparejamiento de letras o monómeros de las secuencias. Para alineamientos de ADN, como hay sólo 4 nucleótidos, se usa una matriz 4x4, que contiene un valor para cada posible emparejamiento de nucleótidos.
|
Además de estas matrices necesitamos un convenio para evaluar de manera adecuada la apertura de gaps en las secuencias que estamos alineando.
Los algoritmos de alineamiento pueden verse como algoritmos de búsqueda, que tratan de encontrar valores óptimos de funciones objetivo que combinan las puntuaciones de las parejas de letras alineadas y el coste de abrir gaps dentro de las secuencias.
Cuando alineamos secuencias enteras hablamos de alineamientos globales ; por el contrario, cuando buscamos sólo alinear las subsecuencias más parecidas, hacemos alineamientos locales .
El alineamiento más sencillo es entre 2 secuencias, en inglés pairwise . Cuando alineamos a la vez más de 2 secuencias hablamos de alineamientos múltiples .
Podemos condensar un alineamiento múltiple de proteínas en un perfil , que es algo así como una matriz de sustitución de aminoácidos que en vez de ser 20x20 toma la forma Nx20, donde N es la longitud del alineamiento.
Además de usar matrices de sustitución para guiar la construcción de un alineamiento podemos usar fuentes de información adicionales. Estas fuentes de información deben poder ser codificadas en forma de secuencia también para que sean útiles.
Básicamente distinguimos entre algoritmos que exploran una fracción grande del espacio de alineamientos, con costes que se aproximan al producto de las longitudes de las secuencias alineadas, y algoritmos que simplifican la búsqueda en ese espacio, que pierden sensibilidad pero requieren menos tiempo y memoria para computar alineamientos.
El primer tipo de algoritmos, más exhaustivos, suelen utilizar programación dinámica para calcular alineamientos. Sirvan de ejemplo los algoritmos de Needleman & Wunsch (1970) y Smith & Waterman (1981) para alineamientos globales y locales, respectivamente, y disponibles aquí y aquí.
En las siguientes secciones veremos ejemplos de algoritmos, y herramientas basadas en ellos, del segundo tipo.
La referencia imprescindible para hacernos una idea de cómo funciona BLAST es Altschul et al. (1997), disponible aquí.
Hay multitud de servidores que corren BLAST en la web para hacer búsquedas de secuencias a través de la red. El padre o la madre de todos ellos es el del NCBI. Estas herramientas son fáciles de utilizar y son a veces más que suficientes para la mayoría de las necesidades, pero si queremos aprovechar todas las posibilidades que nos brinda este paquetes de programas lo mejor es instalarlo localmente.
Los binarios ejecutables de BLAST están disponibles para toda una serie de plataformas y los podemos descargar empaquetados desde aquí.
Por ejemplo, para mi sistema eligiría
blast-2.2.13-ia32-linux.tar.gz y
tras desempaquetarlo con
$ tar xvfz blast-2.2.13-ia32-linux.tar.gz
obtengo un directorio que contiene a su vez:
bin data doc VERSION
En
bin
encontramos los ejecutables binarios, en
doc
la documentación que explica cómo instalar y
usar los programas y en
data
algunos archivos de texto con parámetros e información necesaria para que BLAST
corra, como por ejemplo algunas matrices de sustitución:
# Matrix made by matblas from blosum62.iij # * column uses minimum score # BLOSUM Clustered Scoring Matrix in 1/2 Bit Units # Blocks Database = /data/blocks_5.0/blocks.dat # Cluster Percentage: >= 62 # Entropy = 0.6979, Expected = -0.5209 A R N D C Q E G H I L K M F P S T W Y V B Z X * A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 -1 -4 R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4 N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4 D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4 C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -1 -4 Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4 E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4 H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4 I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4 L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4 K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4 F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4 P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -1 -4 S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 -1 -4 T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 -1 -4 W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -1 -4 Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4 V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4 B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4 Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 X -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -4 * -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1
żQué es una base de datos de secuencias? En este contexto es un conjunto de secuencias que representa un universo dentro del cual buscaremos secuencias similares a nuestra secuencia problema, con el fin de hacer inferencia biológica.
Programas como BLAST asumen que la composición de nuestro universo de secuencias no se desvía de las frecuencias de fondo de aminoácidos o nucleótidos en todas las proteínas conocidas. Estas frecuencias de fondo son importantes para estimar las estadísticas de los alineamientos generados. Si estas frecuencias cambian las estadísticas cambiarán y, por tanto, es posible obtener alineamientos con puntuaciones artificalmente altas o bajas.
Por esta razón es habitual que el primer paso a la hora de preparar una base de datos de secuencias es filtrar las regiones con composiciones atípicas, de baja complejidad (low complexity en inglés) . En ocasiones estas regiones de baja complejidad pueden ser biológicamente de gran interés y deberemos decidir si nos conviene filtrarlas o no. Pero en general es buena idea hacerlo. Aquí usaremos el programa SEG para ello (Wootton & Federhen, 1996), que podéis descargar de aquí .
En cualquier caso, deberemos formatear y preprocesar nuestro conjunto de secuencias para usarlo
como base de datos de secuencias. Podemos usar compilaciones de secuencias estándares, como
las del NCBI, o crear nuestras propias
bases de datos. Para ello deberemos utilizar el programa
formatdb,
incluido en el subdirectorio
bin
de BLAST. Si tecleamos
$ formatdb - el programa nos muestra unas breves instrucciones
de cómo invocarlo.
Este programa permite formatear un archivo de secuencias (en formato FASTA por defecto) y
convertirlo en una base de datos de secuencias contra la que podemos buscar por medio de alineamientos
locales. La sintaxis más sencilla para un conjunto de secuencias de proteína sería:
$ formatdb -i secuencias.fas
donde el contenido de
secuencias.fas
sería algo así como:
>sp|P0ACJ8|CRP_ECOLI Catabolite gene activator Escherichia coli. MVLGKPQTDPTLEWFLSHCHIHKYPSKSTLIHQGEKAETLYYIVKGSVAVLIKDEE... >sp|P29281|CRP_HAEIN Catabolite gene activator Haemophilus influenzae. MSNELTEIDEVVTSSQEEATQRDPVLDWFLTHCHLHKYPAKSTLIHAGEDA... >sp|O05689|CRP_PASMU Catabolite gene activator Pasteurella multocida. MQTTPSIDPTLEWFLSHCHIHKYPSKSTLIHAGEKAETLYYLIKGSVAVLVKDEDGKEMI...
Tras ejecutar $ formatdb -i secuencias.fas obtenemos una serie de archivos que son
los que BLAST usará propiamente para hacer búsquedas contra esta base de datos. Si el archivo
de secuencias es grande entonces esta tarea puede tardar un rato. El trabajo de
formatdb en realidad consiste en convertir las secuencias originales en binario
e indexarlas de forma que al hacer una búsqueda con BLAST se acelere considerablemente
el proceso.
Se pueden hacer varios tipos de búsquedas con BLAST, como podemos ver
aquí, pero las dos
representativos son
blastp
y
blastn,
para hacer búsquedas
de secuencias de proteínas y nucleótidos respectivamente. Todas estas búsquedas se realizan con el mismo ejecutable, llamado
blastall,
que podemos invocar de esta manera para que nos muestre todas sus posible opciones:
$ blastall -
Las opciones más destacadas serían:
blastall 2.2.13 arguments:
-p Program Name [String]
-d Database [String]
default = nr
-i Query File [File In]
default = stdin
-e Expectation value (E) [Real]
default = 10.0
-m alignment view options:
0 = pairwise,
1 = query-anchored showing identities,
2 = query-anchored no identities,
3 = flat query-anchored, show identities,
4 = flat query-anchored, no identities,
5 = query-anchored no identities and blunt ends,
6 = flat query-anchored, no identities and blunt ends,
7 = XML Blast output,
8 = tabular,
9 tabular with comment lines
10 ASN, text
11 ASN, binary [Integer]
default = 0
-o BLAST report Output File [File Out] Optional
default = stdout
-F Filter query sequence (DUST with blastn, SEG with others) [String]
default = T
-v Number of database sequences to show one-line descriptions for (V) [Integer]
default = 500
-b Number of database sequence to show alignments for (B) [Integer]
default = 250
-M Matrix [String]
default = BLOSUM62
Una manera muy sencilla de correr BLAST sería:
$ blastall -p blastp -i misecuencias.fas -d basedesecuencias
De esta manera los resultados de BLAST se muestran en la salida estándar (pantalla), al no especificar
la opción
-o.
En este caso buscamos contra una biblioteca de secuencias llamada
basedesecuencias,
y esa búsqueda se realiza con todas las secuencias (al menos una) que haya, en formato FASTA, dentro de
misecuencias.fas.
Esta búsqueda se hace con todos los parámetros de BLAST con sus valores por defecto.
Por defecto, BLAST muestra una lista de secuencias encontradas de como máximo 500 secuencias (parámetro
-v),
y para cada una de ellas muestra su puntuación en bits y su significancia estadística.
Asimismo, por defecto muestra un máximo de 250 alineamientos (parámetro
-b).
BLAST sólo muestra información de los alineamientos que tengan un valor esperado (en inglés e-value)
igual o menor que un límite predefinido (parámetro
-e),
que por defecto vale 10. Pero, żqué significa este valor? Es el número de secuencias, respecto al tamaño de la
base de datos que estamos usando, que obtendrían la misma puntuación por azar al ser alineadas. Por tanto, cuanto mayor sea
el tamaño de la biblioteca de secuencias que estamos explorando, más significativos serán los valores esperados.
Este valor de expectancia no tiene ninguna interpretación biológica, es una cantidad estadística, y dependiendo de qué inferencias estemos haciendo puede tener diferentes sentidos.
Si la secuencia que estamos usando para hacer una búsqueda BLAST es muy larga, entonces es probable que los alineamientos locales que encuentre BLAST para ella cubran diferentes partes de la secuencia. Una manera visual de observar esto es mediante el programa MVIEW (Brown et al., 1998), que podéis descargar de aquí .
Lo mejor es que llevemos a la práctica estas cosas, con secuencias reales, extraídas de proyectos de secuenciación reales . Os sugiero que juguéis con algunas secuencias y que:
formatdb,
por ejemplo con secuencias de SCOP que podéis tomar de aquí.
De nuevo os sugiero la lectura atenta de Thompson et al. (1994), escrito por los autores del programa CLUSTAL, y disponible aquí.
En breve, CLUSTAL es un programa que permite hacer alineamientos globales de proteínas y ácidos nucleicos y que además tiene un algoritmo heurístico progresivo, bastante rápido, para calcular alineamientos múltiples. En combinación con herramientas como BLAST, CLUSTAL es muy útil para definir familias de proteínas y de ácidos nucléicos.
Al igual que BLAST, también hay servidores web para correr CLUSTALW sin necesidad de instalar software, pero asimismo tiene ventajas instalarlo localmente, sobre todo para correr trabajos de alineamiento múltiple a gran escala y tener todo el proceso bajo control.
Ya tenéis CLUSTALW instalado en el sistema, pero es fácil descargar la última versión aquí.
CLUSTALW es un programa con menús de texto, como podéis comprobar tecleando en un terminal:
$ clustalw
El programa tiene toda una serie de opciones, puede convertir formatos y hacer árboles filogenéticos,
pero de momento aquí veremos su capacidad para hace alineamientos múltiples, la opción 2.
La mejor manera de aprender a usar CLUSTAL es con un ejemplo real. A menudo se hace necesario un alineamiento múltiple con secuencias que hemos seleccionado con BLAST, FASTA u otra herramienta de búsqueda. Precisamente os propongo que:
bl2seq,
del paquete BLAST?
Ahora que hemos visto cómo hacer alineamientos múltiples con CLUSTALW (sería similar con otras herramientas, como T-COFFEE, por ejemplo), estamos ya preparados para aprender a utilizar perfiles de secuencias. Para ello propongo la lectura de este artículo pionero de Gribskov et al. (1987), donde se utilizaron perfiles de secuencias para alinear secuencias muy divergentes.
En breve, definiremos un perfil como una forma condensada de alineamiento múltiple que a su vez puede alinearse con más secuencias. Un perfil consta de una secuencia (de aminoácidos o nucleótidos) donde para posición conocemos las frecuencias de sustitución. Al contrario de cuando usamos matrices de sustitución de propósitos general, como BLOSUM, 2 alaninas en la misma proteína pueden tener diferentes tendencias de sustitución evolutiva. Hay diferentes maneras de implementar perfiles de secuencia, aquí veremos la más sencilla, pero debemos saber que hay otras más complejas, basadas por ejemplo en modelos de Markov, que no sólo incluyen las frecuencias locales de sustitución sino también frecuencias de apertura de gaps en cada posición de la secuencia.
Para entender mejor estas palabras lo mejor es, de nuevo, volver a casos reales. Para ello utilizaremos CLUSTALX, la versión gráfica de CLUSTAL, que ya tenéis instalada en vuestro sistema, y PSI-BLAST, la versión iterativa de BLAST :
blastpgpg
con alguna de las secuencias iniciales, usando 3 iteraciones, contra la base de secuencias SCOP. żCómo crees que utiliza PSI-BLAST los perfiles?
blastgp?
żQué es el archivo que genera?
Este artículo de Eddy (2004) explica en qué consisten los modelos ocultos de Markov y su utilidad en biología.
En clase veremos cómo usar HMMs por medio de ejemplos, que viven en /home/bioinfo/LCG/hmmer-tutorial/
y deberéis copiar a vuestros propios directorios. Para ellos usaremos el paquete de programas
HMMER, desarrollado en el grupo de Sean Eddy.
globins50.msf, pero HMMER puede leer diferentes formatos: $ hmmbuild globins.hmm globins50.msf
$ hmmcalibrate globins.hmm
hmmemit -n 5 globins.hmm
$ hmmsearch globins.hmm Artemia.fa
$ hmmalign -o globins630.ali globins.hmm globins630.fas
$ hmmsearch globins.hmm /home/bioinfo/LCG/FAA_Genome/Z_mobilis_ZM4.faa
$ hmmbuild rrm.hmm rrm.sto $ hmmbuild fn3.hmm fn3.sto $ hmmbuild pkinase.hmm pkinase.sto $ cat rrm.hmm fn3.hmm pkinase.hmm > libreria.hmm $ hmmcalibrate libreria.hmm
$ hmmpfam varios.hmm 7LES_DROME
Espero que a estas alturas ya esté claro qué es un perfil y qué diferencia PSI-BLAST de BLAST. Aprovechando que tenéis ya cierta práctica en la programación se pide que:
Ya vimos en el capítulo 4 qué herramientas bioinformáticas podemos utilizar para manipular secuencias biológicas. En este capítulo vamos más allá de las secuencias, vamos a tratar de caracterizar macromoléculas biológicas con ayuda de la bioinformática. Para empezar, con la idea de conectar el tema anterior con este, veremos que hay ciertas propiedades moleculares que pueden estudiarse conociendo sólo la secuencia de la molécula de interés.
El artículo de Kanhere & Bansal (2005), disponible aquí, presenta un método de predicción de regiones promotoras en bacterias que se basa en el cálculo de la estabilidad helicoidal del ADN cromosómico en tales regiones. Sin saber muchos de los detalles moleculares del proceso de expresión génica, este trabajo sugiere que un gen, para ser expresado, requiere que el complejo de transcripción abra la doble hélice de ADN.
Para acercarnos a lo que hicieron Kanhere & Bansal (2005) podemos utilizar el programa calculateGibbs.pl, escrito en Perl, y el archivo K12_400_50_sites, que contiene las secuencias promotoras de los genes de Escherichia coli de las que conocemos al menos un sitio de pegado de factores de transcripción. Los ejercicios propuestos son:
La reacción en cadena de la polimerasa tiene muchas aplicaciones en biología, medicina y biotecnología. Todas estas aplicaciones dependen del empleo de parejas de cebadores o primers, oligonucleótidos que sirven de puntos de arranque de la actividad polimerasa. En concreto, la extensión de los cebadores se da en su extremo 3'. Por estas razones es deseable que los cebadores usados en una PCR cumplan ciertas condiciones, todas ellas calculables desde su secuencia:
En la Web hay ejercicios para aprender a diseńar primers como éste y éste otro.
El artículo de Rose et al. (2003), disponible aquí, presenta una de las aplicaciones del diseńo de primers degenerados.
La tarea de hoy incluye:
La estructura secundaria es una propiedad estructural de las proteínas que puede predecirse con bastante precisión a partir de su secuencia. Hay una gran variedad de algoritmos y aproximaciones válidas para este problema, pero todos ellos suelen predecir para cada residuo un estado seleccionado de un repertorio de tres: hélice alfa, lámina beta y algo así como lazo (coil en inglés. El conocimiento de la estructura secundaria de una proteína puede tener muchas aplicaciones a la hora de diseñar experimentos y a la hora de diseñar algoritmos que predicen la estructura terciaria de una proteína.
Veamos una buena exposición de este tema en este artículo de Jones (1999), donde se presenta la herramienta PSIPRED y sus algoritmos subyacentes.
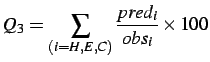 |
(5.1) |
 |
(5.2) |
En la sección anterior vimos un método que permite reconocer regiones estructuradas dentro de la secuencia de una proteína. En esta sección veremos una herramienta complementaria, llamada DISOPRED, que está basada en la anterior, que además de reconocer elementos de estructura secundaria, calcula la probabilidad empírica de que cada aminoácido de la proteína este desordenado, sin pertenecer a ninguna clase de estructura secundaria, en la forma biológicamente activa de una proteína.
Ahora, como ejercicio, la idea es aprender a utilizar DISOPRED, disponible en
/home/bioinfo/psipred/:
rundisopred
y comprender sus tripas
rundisopred en sus búsquedas?
rundisopred
a tu home y modificarlo convenientemente?
La estructura secundaria de las moléculas de RNA, sostenida por medio de puentes de hidrógeno entre nucleótidos, puede aproximarse a partir del conocimiento de la secuencia. Una de sus principales aplicaciones es el descubrimiento de RNAs no codificantes, que pueden tener funciones biológicas importantes, como se discute en este artículo de Washietl et al. (2005).
En esta clase la idea es explorar el paquete de programas Vienna para estudiar la estructura secundaria de moléculas de RNA (puede ser útil revisar este tutorial para recordar la teoría). Se sugiere para el trabajo en clase trabajar con dos ejemplos:
Cómo mencionamos en la sección 4.1, el uso de secuencias nos permite simplificar la manipulación y la inferencia con moléculas biológicas. Sin embargo, no todas las propiedades moleculares pueden ser capturadas por una sola secuencias de caracteres ni pueden ser calculadas a partir de ella.
En las siguientes secciones utilizaremos el formato PDB ( Protein Data Bank), que ya tocamos en la sección anterior, para codificar la estructura tridimensional de macromoléculas biológicas, sobre todo proteínas y ácidos nucléicos, por medio de las coordenadas cartesianas de sus átomos, deducidas experimentalmente.
Empezaremos por ver qué relación hay entre la secuencia y la estructura de las proteínas, leyendo este artículo fundamental de Chothia & Lesk (1986). Que yo sepa no hay un trabajo similar hecho con los ácidos nucleicos, quizás porque el número de ácidos nucleicos de los que conocemos su estructura es menor y porque con el ADN, al ser helicoidal, no tiene tanto interés.
Una vez entendido el concepto de correlación entre similitud a nivel de secuencia y a nivel de estructura, vamos a trabajar con el programa MAMMOTH (Ortiz et al., 2002):
Con las herramientas que hemos estado manejando hasta ahora ya estamos en disposición de modelar proteínas, si nos surgiera esa necesidad. Por modelar quiero decir hacer una predicción de cómo se disponen los átomos de una proteína conocida su secuencia. Las aplicaciones que puede tener un modelo son amplias, desde la biología molecular al diseño de fármacos, pasando por la inferencia evolutiva y funcional.
En esta sección veremos que en ocasiones podemos asumir que el esqueleto peptídico de una proteína (en inglés backbone) no va a variar mucho al cambiar su secuencia. Esto puede tener aplicaciones como la que vemos en este artículo de Reina et al. (2002), donde se rediseña una proteína para cambiar su afinidad por un ligando.
Como ejercicio para poner estas cosas en práctica sugiero aprender a utilizar el programa SCWRL, diseñado por Bower et al. (1997), que encontraréis en /home/bioinfo/scwrl. SCWRL, que en inglés se pronuncia algo así como 'ardilla', calcula las cadenas laterales de una proteína dada la geometría de su esqueleto. Para ello utiliza una librería de cadenas laterales precompiladas, extraídas del Protein Data Bank, cuyas orientaciones (rotámeros) dependen de la geometría local del esqueleto:
-s
En la sección anterior ya hemos modelado proteínas y péptidos, pero sin tocar el esqueleto peptídico. En ésta vamos más allá y veremos un protocolo general de modelado comparativo de proteínas, el que se usa cuando no conocemos una estructura molecular pero sí la de proteínas similares en secuencia.
Para ello empezamos por este artículo de (Fiser & Sali, 2003), disponible aquí. Otros artículos relevantes en este campo podrían ser (Contreras-Moreira et al., 2005) y (Zhang & Skolnick, 2005).
El ejercicio de hoy implica el programa MODELLER
(Sali & Blundell, 1993), disponible sin costo para usuarios académicos, que ya está instalado en el
sistema como
/home/bioinfo/bin/modeller8v1/bin/mod8v1
. El programa tiene múltiples opciones y utilidades,
aquí veremos su uso fundamental para construir modelos comparativos. Vamos a hacerlo paso a paso:
.ali:
C; A sample alignment in the PIR format; used in tutorial >P1;5fd1 structureX:5fd1:1 : :106 : :ferredoxin:Azotobacter vinelandii: 1.90: 0.19 AFVVTDNCIKCKYTDCVEVCPVDCFYEGPNFLVIHPDECIDCALCEPECPAQAIFSEDEVPEDMQEFIQLNAELA EVWPNITEKKDPLPDAEDWDGVKGKLQHLER* >P1;1fdx sequence:1fdx:1 : :54 : :ferredoxin:Peptococcus aerogenes: 2.00:-1.00 AYVINDSC--IACGACKPECPVNIIQGS--IYAIDADSCIDCGSCASVCPVGAPNPED----------------- -------------------------------*
.py),
como éste:
# Homology modelling by the automodel class
from modeller.automodel import * # Load the automodel class
log.verbose() # request verbose output
env = environ() # create a new MODELLER environment to build this model in
# directories for input atom files
env.io.atom_files_directory = './'
a = automodel(env,
alnfile = 'alignment.ali', # alignment filename
knowns = '5fd1', # codes of the templates
sequence = '1fdx') # code of the target
a.starting_model= 1 # index of the first model
a.ending_model = 1 # index of the last model
# (determines how many models to calculate)
a.make() # do the actual homology modelling
/home/bioinfo/bin/modeller8v1/bin/mod8v1
En ocasiones una búsqueda de secuencias con BLAST u otra herramienta similar no es suficiente para encontrar estructuras molde para el modelado. En esos casos podemos probar a hacer una búsqueda más sofisticada, de reconocimiento de plegamientos (fold recognition o threading en inglés). Herramientas como BIOINFO.PL:META pueden a veces encontrar moldes adecuados para el modelado.
Esto es ya más avanzado, no lo veremos en clase, pero debéis saber que hay disponibles herramientas para modelar proteínas sin moldes, sólo desde su secuencia, y que con ciertas restricciones son de gran utilidad. Os recomiendo ROBETTA (Kim et al., 2004), disponible como servidor web y también como código fuente, previa firma de una licencia.
En ocasiones puede ser de interés modelar moléculas de DNA de doble hélice, como vimos previamente a la hora de localizar promotores en genomas, como se discute en este artículo de Michael Gromiha et al. (2004), para estudiar el modo de reconocimiento de secuencias de DNA por parte de factores de transcripción.
En esta clase usaremos el paquete de programas X3DNA, disponible en forma de binarios para diferentes arquitecturas, con el fin de analizar la conformación de moléculas de DNA (resueltas por cristalografía on NMR) y de modelar secuencias de acuerdo a conformaciones conocidas del esqueleto helicoidal.
Las tareas requeridas para esta clase son:
X3DNA/Examples/Analyze_Rebuild/README con tu molécula de DNA.
Existen servidores web como MDDNA o READOUT que pueden ser útiles en estas tareas.
Después de las últimas sesiones creo que ya estáis preparados para un ejercicio de modelado real. En concreto os pido que:
Aquí iré poniendo enlaces relacionados con este curso y los que me vayáis enviando vosotros:
An introduction to bioinformatics algorithms
This document was generated using the LaTeX2HTML translator Version 2002-2-1 (1.70)
Copyright © 1993, 1994, 1995, 1996,
Nikos Drakos,
Computer Based Learning Unit, University of Leeds.
Copyright © 1997, 1998, 1999,
Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The command line arguments were:
latex2html bioinfo -split 0 -dir bioinfo1 -no_navigation
The translation was initiated by Bruno Contreras Moreira on 2007-05-01
![\begin{figure}\begin{center}
\includegraphics[width=0.8\textwidth]{clusterreal}\end{center}\end{figure}](img2.png)